The Two Records
Evolution left two kinds of evidence. The first is the fossil record: the preserved remains of organisms that lived and died across geological time, layered in rock strata in a sequence that traces the history of life from its earliest single-celled forms to the present. The fossil record is a magnificent archive, but it is incomplete in ways that are not random. Hard structures, bones, shells, teeth, preserve far better than soft tissue. Small organisms preserve less reliably than large ones. Environments that permit fossilisation represent a tiny fraction of the environments in which life has actually existed. Enormous spans of evolutionary history have left almost no direct fossil trace.
The second record is the molecular record: the information stored in the genomes of living organisms. Every living cell carries within it a document written in nucleotides that records, in molecular shorthand, much of the evolutionary history of its lineage. The substitutions that have accumulated in a gene since two lineages diverged are a function of the time since divergence and the rate at which mutations arise. The architecture of a genome records the major transitions in the history of the lineage. The molecular record is not a replacement for the fossil record. It is a different kind of evidence, capable of answering questions the fossil record cannot reach.
What molecular evolution has produced, over the decades since DNA sequencing became possible, is a transformation in the precision and resolution with which evolutionary history can be reconstructed. It has confirmed many relationships suggested by morphology and overturned others. It has extended evolutionary analysis to organisms that leave no fossils at all, including most bacteria and archaea. And it has revealed, at the level of individual nucleotides, the actual mechanisms by which evolution operates: mutation, selection, genetic drift, and gene flow, acting on the raw material of sequence variation in populations of molecules.
Mutation Again, This Time as Evolutionary Mechanism
In the previous artifact, mutation appeared as a source of disease: the single nucleotide change that causes sickle cell anaemia, the three-base deletion that causes cystic fibrosis. At the level of an individual, most mutations are neutral or harmful. At the level of a population across evolutionary time, mutation is the sole source of the genetic variation on which natural selection acts. Without mutation there is no variation. Without variation there is no selection. Without selection there is no adaptation. The same molecular process that occasionally produces a devastating disease is the engine of all biological innovation in the history of life.
The rate at which mutations arise varies across the genome. Certain regions, including the microsatellite sequences consisting of short repeated motifs, accumulate mutations at rates 1,000 to 10,000 times higher than the genome average, because the replication machinery tends to slip on repeated sequences. Other regions are extraordinarily conserved: some sequences in the human genome are essentially identical to their counterparts in fish and fruit flies, a conservation that reflects not the absence of mutation but the action of purifying selection removing mutations as fast as they arise because the sequence is functionally critical.
The neutral theory of molecular evolution, proposed by Motoo Kimura in 1968, provided the theoretical framework for understanding the molecular record. Kimura argued that the vast majority of mutations that reach high frequency in a population do so not because they are favoured by selection but because of chance, the random process of genetic drift in finite populations. Most variation in the genome is selectively neutral: it makes no difference to survival or reproduction whether a given position carries an A or a T, and so the fate of variants at such positions is determined by the lottery of inheritance across generations rather than by selection. This was controversial when Kimura proposed it, in a field where selection had been the primary explanatory framework. The molecular data supported his view. Synonymous substitutions, which do not change the amino acid sequence of a protein and are therefore invisible to selection, accumulate faster than non-synonymous substitutions that do change amino acids, exactly as neutral theory predicts.
The neutral theory does not displace natural selection. It provides the null model against which selection can be detected. When a genomic region shows a pattern of substitution that deviates significantly from the neutral expectation, selection is operating. Regions that have accumulated far fewer substitutions than neutrally expected are under purifying selection, removing harmful mutations. Regions that have accumulated more non-synonymous than synonymous substitutions are under positive selection, with beneficial mutations being actively favoured. These signatures in the sequence are readable decades or millions of years after the selection occurred.
The Molecular Clock
One of the most powerful tools that molecular evolution has produced is the molecular clock: the observation that some categories of molecular change accumulate at approximately constant rates over evolutionary time, allowing the timing of evolutionary events to be estimated from the amount of molecular divergence between lineages.
The molecular clock was proposed empirically by Emile Zuckerkandl and Linus Pauling in 1965, based on their observation that the amino acid differences in haemoglobin between pairs of species were roughly proportional to the time since those species diverged, as estimated from the fossil record. If haemoglobin changed at a roughly constant rate, then the amount of haemoglobin difference between two species could be used to estimate when they last shared a common ancestor, independent of any fossil evidence.
The theoretical basis of the molecular clock is provided by neutral theory. If most substitutions are neutral and accumulate at a rate determined by the mutation rate rather than by selection, then the substitution rate should be roughly constant across evolutionary time and across lineages. The mutation rate does vary somewhat between lineages and between genomic regions, which is why the clock is sometimes described as approximately constant rather than perfectly regular. But for many genes and many comparisons, the molecular clock provides a reliable dating tool with accuracy sufficient to resolve major questions about the timing of evolutionary events.
The molecular clock has been applied to some of the most consequential questions in human evolutionary history. The divergence time between humans and chimpanzees has been estimated from molecular data at approximately 5 to 7 million years ago, consistent with the fossil record for early hominins. The divergence between Homo sapiens and Neanderthals has been estimated at approximately 600,000 to 800,000 years ago. The split between anatomically modern humans and all other human populations has been traced to Africa approximately 200,000 to 300,000 years ago, with subsequent migrations out of Africa beginning approximately 60,000 to 70,000 years ago.
The molecular clock has also been decisive in questions where the fossil record is silent. The last common ancestor of all living humans, the mitochondrial Eve, has been placed at approximately 150,000 to 200,000 years ago using the mutation rate of mitochondrial DNA. Mitochondrial Eve is not the first woman, or the only woman alive at the time. She is defined as the most recent woman from whom all living humans descend through an unbroken female line. The mitochondrial lineage traces a single thread of female descent through an enormous, branching genealogy.
Chart 01
Primate Divergence Times Estimated from Molecular Clock Data
Natural Selection at the Molecular Level
The evidence that natural selection has operated on specific genes and genomic regions in the human lineage is now extensive and specific. Genomic methods allow the identification of regions that show the statistical signatures of recent positive selection: extended haplotype blocks, where a new beneficial mutation has swept to high frequency in a population so recently that the surrounding genomic region has not yet had time to be broken up by recombination, reduced genetic diversity around the selected site, and elevated differentiation between populations.
One of the most studied examples of positive selection in the human genome is the lactase persistence allele. In most mammals, the enzyme lactase, which breaks down the milk sugar lactose, is expressed in the gut during infancy and its expression is substantially reduced after weaning. Most humans globally are lactase non-persistent: they lose the ability to digest lactose efficiently in adulthood. But populations with long histories of pastoralism, particularly in northern Europe and certain East African and Arabian pastoral groups, have high frequencies of genetic variants that maintain lactase expression throughout adult life. These variants arose independently in different populations and show some of the strongest signatures of recent positive selection in the human genome. The estimated age of the European lactase persistence allele is approximately 5,000 to 10,000 years ago, making it one of the most recent strong selection events identified in the human genome.
The evolution of the human brain has left distinctive molecular signatures. The gene ASPM, which when mutated causes microcephaly, shows evidence of positive selection in the lineage leading to humans, with an unusually high ratio of non-synonymous to synonymous substitutions compared to its orthologues in other primates. The gene FOXP2, associated with the neural circuitry underlying speech and language, shows two amino acid changes in the human lineage that are not present in any other primate, changes that show evidence of positive selection. Wolfgang Enard and colleagues at the Max Planck Institute demonstrated in 2002 that these human-specific changes in FOXP2 were fixed in the human population more recently than would be expected under neutral evolution, consistent with selection having driven them to fixation within the last 200,000 years.
The amylase gene AMY1 has undergone a dramatic expansion in copy number in the human genome relative to other primates. Most humans carry between 2 and 15 copies of AMY1 per haploid genome, compared to 2 copies in chimpanzees. AMY1 encodes salivary amylase, the enzyme that begins starch digestion in the mouth. Populations with traditionally high-starch diets have higher average AMY1 copy numbers than populations with lower-starch diets, suggesting that AMY1 copy number expansion was selected for as human ancestors shifted toward starchy plant foods. A dietary shift tens of thousands of years ago is recorded in the structure of the human genome today.
Genetic Drift and the Importance of Chance
Natural selection is the mechanism that produces adaptation, but it is not the only evolutionary force shaping the genome. Genetic drift, the random change in allele frequencies that occurs in finite populations simply because each generation inherits a random sample of the parental generation's gametes, has been a major sculptor of human genetic diversity.
The importance of drift relative to selection depends on population size. In very large populations, selection is efficient even for weakly beneficial mutations, because each new mutation is present in enough individuals that chance fluctuations rarely eliminate it before selection can act. In very small populations, chance fluctuations can overwhelm selection: a mildly beneficial mutation may be lost simply because the few individuals who carry it happen not to reproduce, and a mildly harmful mutation may rise to fixation simply because the individuals who carry it happen to leave more offspring in one generation. Kimura's neutral theory formalised this relationship: a mutation is effectively neutral if its selective advantage or disadvantage is smaller than the inverse of the effective population size.
The human species has gone through severe population bottlenecks during its history, episodes in which the total population was reduced to a small number of individuals, greatly amplifying the effects of drift. The expansion of modern humans out of Africa appears to have involved a dramatic reduction in population size: non-African populations show consistently lower genetic diversity than African populations, a signature of the founder effect associated with small groups of migrants founding the populations that expanded across Eurasia, the Americas, and the Pacific.
The founder effect has medical consequences. Certain genetic diseases reach unusually high frequencies in populations founded by small groups in which a carrier happened to be present. Tay-Sachs disease is more frequent in Ashkenazi Jewish populations than in most others. Certain forms of familial hypercholesterolaemia are unusually common in Afrikaner populations in South Africa, traceable to a founder effect in the original Dutch settler community. These elevated frequencies are not the result of selection maintaining a harmful allele. They are the result of chance: a small founding population happened to include carriers, and the allele frequency was amplified by the random sampling process that produced subsequent generations.
Gene flow, the movement of alleles between populations through migration and interbreeding, counters the divergence that selection and drift produce. The Neanderthal and Denisovan sequences present in modern human genomes are the molecular evidence of ancient gene flow events. Some of the sequences acquired through these events appear to have been subsequently favoured by selection, conferring adaptations to local environments, including high-altitude adaptation in Tibetan populations from Denisovan-derived sequence at the EPAS1 locus.
Phylogenomics and the Tree of Life
Before molecular data became available, the relationships between organisms were inferred from comparative anatomy, embryology, and the fossil record. These methods produced a broadly correct picture of the major branches of the tree of life, but left many relationships unresolved or incorrectly placed, particularly for organisms whose morphology was simple or convergent.
Molecular phylogenetics, and its successor phylogenomics, which uses whole-genome data rather than single genes, has produced a resolution of the tree of life that was unattainable by morphological methods. The approach is conceptually simple: closely related species share more molecular similarity than distantly related ones, and the pattern of similarity across many genes or many positions in the genome can be used to reconstruct the branching order of lineages with statistical confidence.
Carl Woese at the University of Illinois performed the analysis that restructured the entire understanding of the living world. In 1977, using ribosomal RNA sequences as phylogenetic markers, he showed that what had been classified as bacteria actually comprised two profoundly distinct groups: the true bacteria, or Eubacteria, and a group he named the Archaea, which had been hidden among the bacteria in classification systems that could not see their molecular distinctiveness. The archaea are as different from bacteria at the molecular level as either is from eukaryotes. Woese's 1977 paper reorganised the tree of life at its deepest level, replacing the two-domain classification with a three-domain tree: Bacteria, Archaea, and Eukarya.
Molecular phylogenetics has also resolved long-standing disputes about the relationships between living mammals. The grouping of cetaceans, whales, dolphins, and porpoises, as the closest living relatives of hippopotamuses, nested within the artiodactyls, was first strongly supported by molecular data in the 1990s and has since been confirmed by multiple genomic analyses. The morphological evidence had pointed elsewhere: hippos had been grouped with pigs based on shared anatomical features that are now understood to be primitive retentions rather than shared derived characters. The molecule read the relationship that the bone could not.
Horizontal gene transfer (HGT), the movement of genes between organisms other than through reproduction, complicates the picture of a simple tree of life in the microbial world. Bacteria and archaea transfer genes between lineages at rates that make the reconstruction of their phylogeny more like a network than a tree. Antibiotic resistance genes spread between bacterial species through HGT, which is why antibiotic resistance can emerge in one species and spread rapidly through entire microbial communities. The tree of life, at the microbial level, is more accurately described as a web.
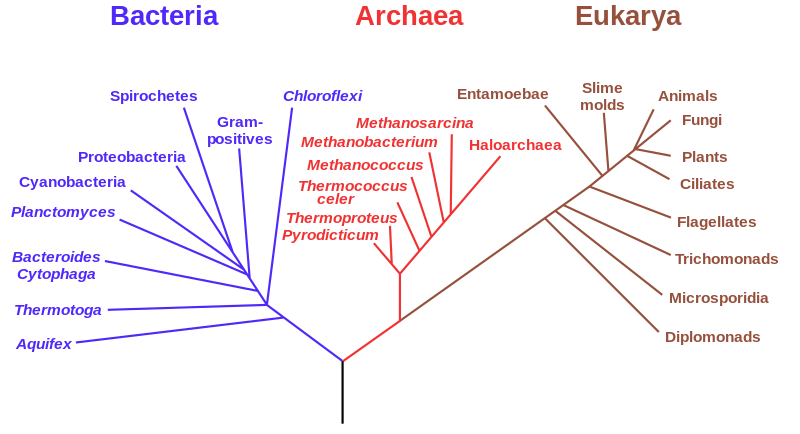
The three-domain tree of life as established by Carl Woese using ribosomal RNA sequence comparisons, first published in 1977. The Archaea, previously classified as unusual bacteria, are revealed as a separate domain as distinct from bacteria as either is from eukaryotes. The eukaryotic domain, which includes all animals, plants, fungi, and protists, appears as a relatively recent branch on a tree whose deepest divisions are microbial.
The Ancestral Genome and What Was Lost
Comparative genomics has made it possible to reconstruct features of ancestral genomes that existed hundreds of millions of years ago, by identifying sequences that are conserved across multiple lineages and inferring the state of the ancestral sequence from which they all descend.
Conserved non-coding elements (CNEs) are genomic sequences that show levels of conservation across distantly related vertebrates far exceeding what would be expected if they were evolving neutrally, despite not encoding any protein. The ENCODE project, a large-scale international effort to characterise the functional elements of the human genome, found that a substantial fraction of conserved non-coding sequences correspond to regulatory elements: enhancers, silencers, and insulators that control when and where genes are expressed during development. These regulatory sequences are often as critical to normal development as the protein-coding sequences they control.
The human genome is littered with the evidence of genes that existed in ancestors but were lost in the human lineage. The olfactory receptor gene family is the largest gene family in the mammalian genome; mice have approximately 1,300 functional olfactory receptor genes, while humans have only approximately 400, with the remaining 300 to 400 olfactory receptor-like sequences being pseudogenes: structurally present but functionally silenced by inactivating mutations. The reduction in the human olfactory receptor repertoire corresponds to the evolutionary shift toward visual dominance in the primate lineage.
The gene for GULO (L-gulonolactone oxidase), required for vitamin C synthesis, is present in the human genome as a pseudogene, carrying a set of inactivating mutations that appear to have accumulated after the ancestor of the primate lineage shifted to a diet sufficiently rich in vitamin C from fruit that endogenous synthesis was no longer necessary. The pseudogene is a molecular tombstone: the precise location in the genome where a functional capability died.
Svante Paabo and the Ancient Genome
The reconstruction of evolutionary history from living genomes has been transformed by the development of ancient DNA analysis, the extraction and sequencing of DNA from fossilised or preserved remains of organisms that died thousands or hundreds of thousands of years ago.
Svante Paabo at the Max Planck Institute for Evolutionary Anthropology in Leipzig has been the central figure in this field since its inception. His early work in the 1980s and 1990s demonstrated that DNA could be recovered from ancient specimens despite its tendency to degrade over time into short, chemically damaged fragments. The technical challenges are severe: ancient DNA molecules are highly fragmented, chemically modified in characteristic ways that cause sequencing errors if not corrected for, and typically present in tiny amounts overwhelmed by contaminating DNA from bacteria, fungi, and modern human handlers. Paabo's group developed the laboratory protocols and bioinformatic methods to address each of these challenges.
The landmark achievement of Paabo's group was the sequencing of the complete Neanderthal genome, published in Science in 2010. The source material was bone from three Neanderthal individuals from Vindija Cave in Croatia, approximately 38,000 to 44,000 years old. The genome was assembled from millions of short, damaged ancient DNA fragments and then compared to the reference human genome. The comparison showed that non-African modern humans carry approximately 1 to 4 percent of their genome from Neanderthal ancestors, direct evidence of interbreeding between anatomically modern humans and Neanderthals during or shortly after the migration of modern humans out of Africa.
In 2010, Paabo's group also published the genome of the Denisovans, derived from a single finger bone fragment found in Denisova Cave in Siberia. The fragment was small enough to fit in a palm, but the DNA preserved within it was of exceptional quality, permitting a genome reconstruction accurate enough to identify the Denisovans as a hominin population distinct from both modern humans and Neanderthals, with a divergence time from the Neanderthal lineage of approximately 400,000 years ago. An entire human population, previously unknown, was identified from the DNA in a single finger bone. No skull, no postcranial skeleton. A genome, and a position on the tree of life, derived entirely from nucleotide sequence.
Paabo received the Nobel Prize in Physiology or Medicine in 2022 for his discoveries concerning the genomes of extinct hominins and human evolution.

Denisova Cave in the Altai mountains of Siberia, where in 2008 a fragment of a finger bone was recovered from sediments dating to approximately 40,000 to 50,000 years ago. The DNA extracted from this fragment, sequenced by Svante Paabo's group, revealed an entirely unknown hominin population. The Denisovans are now known to have interbred with the ancestors of present-day Melanesian and Aboriginal Australian populations, contributing 4 to 6 percent of their genomes.
Gene Duplication and Innovation
Evolution requires not only changes in existing genes but the creation of new genes with new functions. One of the primary mechanisms by which new genes arise is gene duplication: the copying of an existing gene, producing two copies in the genome, after which the two copies are free to diverge in sequence independently. If one copy maintains the original function, the other is relieved of selective constraint and can accumulate mutations that may eventually produce a new function. This is neofunctionalisation: the evolution of a new role for a duplicated gene.
The haemoglobin gene family is one of the best-studied examples of gene duplication and divergence in vertebrate evolution. The ancestral haemoglobin gene duplicated and reduplicated over hundreds of millions of years, producing a family of related genes whose products are specialised for different oxygen-binding roles in different tissues and at different stages of development. Foetal haemoglobin has a higher oxygen affinity than adult haemoglobin, allowing it to extract oxygen from the maternal blood supply across the placenta. The genes encoding foetal haemoglobin are expressed during development and then switched off as adult haemoglobin genes are activated after birth. This developmental programme of haemoglobin gene switching is the product of hundreds of millions of years of gene duplication and divergence.
Whole genome duplication (WGD), events in which the entire genome is duplicated rather than individual genes, have occurred repeatedly in the history of eukaryotic life. Two rounds of WGD are inferred to have occurred in the ancestor of vertebrates, approximately 500 to 600 million years ago, based on the presence in vertebrate genomes of multiple related genes for which only single copies exist in invertebrate genomes. These duplications may have provided the genomic raw material for the evolutionary innovations associated with the origin of vertebrates, including the vertebrate immune system, the complex nervous system, and the diversification of developmental gene families. Flowering plants have undergone WGD repeatedly during their evolutionary history; the genome of bread wheat contains six copies of most genes, the result of three successive rounds of hybridisation and polyploidisation events in the ancestry of the modern wheat lineage.
The Molecular Signature of Selection on the Human Lineage
The comparison of the human genome with the genomes of our closest relatives has made it possible to identify the specific genomic changes that occurred in the human lineage after its divergence from the chimpanzee lineage approximately 6 million years ago.
Katherine Pollard and colleagues identified in 2006 a category of genomic sequences they designated Human Accelerated Regions (HARs): non-coding sequences that are highly conserved across vertebrates generally but show a dramatically elevated rate of change in the human lineage specifically. The most rapidly evolving of these, HAR1, shows 18 substitutions in the human lineage compared to only 2 in the preceding 300 million years of vertebrate evolution. HAR1 is expressed in the developing human cortex during the period of cortical neuron development, in Cajal-Retzius neurons that play a critical role in establishing the layered organisation of the cortex.
Gene losses specific to the human lineage have also attracted attention. The gene MYH16, encoding a myosin heavy chain expressed specifically in jaw muscles, was inactivated by a frameshift mutation in the human lineage approximately 2.4 million years ago, a timing that corresponds roughly to the period of brain size increase in the hominin fossil record. Hansell Stedman and colleagues proposed in 2004 that the loss of MYH16 reduced the mechanical constraints imposed on the skull by powerful jaw muscles, permitting the cranial expansion that accommodated the enlarging brain. A gene's loss, not gain, as a potential driver of a major evolutionary transition.
The emerging picture from comparative genomics is that the human genome differs from the chimpanzee genome not primarily in having acquired many new genes with new functions, but in having changed the regulation of existing genes: where they are expressed, when during development they are switched on and off, and at what levels. Most of the approximately 35 million single nucleotide differences and the 5 million insertions and deletions between the human and chimpanzee genomes fall in non-coding regulatory regions. The human brain is not built from different proteins than the chimpanzee brain. It is built from many of the same proteins, expressed in different patterns, at different times, in different quantities.
Chart 02
dN/dS Ratios: Reading the Signature of Selection from Sequence Alone
What Molecular Evolution Reveals About the Nature of Life
The molecular record of evolution delivers a picture of life that is at once more precise and more strange than the one available before genomics existed.
It is more precise because it is quantitative. Evolutionary relationships that were previously inferred from bone and shell can now be measured in nucleotide substitutions per site per million years. The divergence of two lineages is no longer an approximate date derived from stratigraphy but a statistical estimate with confidence intervals derived from the rates at which molecules change. The degree of relatedness between a human and a chimpanzee, a human and a mouse, a human and a yeast, is a number: a number of shared derived molecular characters that reflects shared evolutionary history with a precision that morphology cannot approach.
It is more strange because the molecular record makes unambiguous what evolutionary theory had always implied but what remained deniable as long as the evidence was primarily bones and stones: that every living thing on Earth is related to every other living thing through common descent, not metaphorically but materially, through an unbroken lineage of replicating molecules stretching back to the origin of life. The 31 percent of the human genome that is recognisably similar to the genome of brewer's yeast is not a coincidence or an artefact. It is the molecular echo of a common ancestor that lived over a billion years ago and passed its molecular systems to all its descendants, who are still using them.
It is not a blueprint for a human being. It is a history of everything that made a human being possible.